Software
![]()
![]()
Software
HPC2N has a non-exclusive, non-commercial use license for academic purposes.
NAMD Molecular Dynamics Software is one of the fastest and highly scalable packages in the world for the simulation of molecular systems.
NAMD is currently being developed by theTheoretical and Computational Biophysics Group at University of Illinois, USA. Together with VMD, its partner software for molecular visualization and simulation setup, NAMD constitutes an essential tool for molecular modellers. NAMD is a flexible software which allows the users to interact with data structures through Tcl or Python scripts. It supports input files from other packages such as CHARMM, and X-PLOR and parameter files from them as well.
NAMD is distributed either as source code or in binary executables for standard arquitechtures including shared memory (SMP), MPI, and CUDA (all these available on our systems).
Besides the basic algorithms for classical molecular dynamics simulations (thermostats, barostats, precise long-range electrostatic methods, etc.), NAMD offers advanced algorithms for enhanced sampling simulations (replica exchange as an example) or free energy computations.
On HPC2N we have NAMD available as a module on Kebnekaise.
Users can find the installed versions of NAMD by typing:
module spider namd
the user will be then offered the following versions of NAMD (subject to change - check with ml spider):
NAMD/2.12-mpi NAMD/2.12-nompi
the first option (mpi) refers to the MPI-Shared memory (SMP) version which can run on multiple nodes and can make use of worker threads on each node (process). The second option (nompi) is the multicore version which can only run on a single node, but because it makes use of local memory it is faster than the MPI-SMP version in this case. Thus, we recommend the nompi version if you are planning to use a single node.
Note: currently, only the nompi option supports the GPU version of NAMD in our system.
Here we present a description of our recommendation on how to run NAMD depending on the version and the number of cores you want to use:
#!/bin/bash #SBATCH -A SNICXXXX-Y-ZZ #Asking for 10 min. #SBATCH -t 00:10:00 #Number of nodes #SBATCH -N 1 #Ask for 8 processes #SBATCH -c 8 #Load modules necessary for running NAMD module add icc/2017.1.132-GCC-6.3.0-2.27 impi/2017.1.132 module add NAMD/2.12-nompi #Execute NAMD namd2 +setcpuaffinity +p6 config_file > output_file
#!/bin/bash #SBATCH -A SNICXXXX-Y-ZZ #Asking for 10 min. #SBATCH -t 00:10:00 #Number of nodes #SBATCH -N 1 #Ask for 28 processes #SBATCH -c 28 #Load modules necessary for running NAMD module add icc/2017.1.132-GCC-6.3.0-2.27 impi/2017.1.132 module add NAMD/2.12-nompi #Execute NAMD namd2 +setcpuaffinity +p28 config_file > output_file
#!/bin/bash #SBATCH -A SNICXXXX-Y-ZZ #Asking for 10 min. #SBATCH -t 00:10:00 #Number of nodes #SBATCH -N 1 #Ask for 28 processes #SBATCH -c 28 #SBATCH --exclusive #Ask for 2 GPU cards #SBATCH --gres=gpu:k80:2 #Load modules necessary for running NAMD module add GCC/5.4.0-2.26 CUDA/8.0.61_375.26 OpenMPI/2.0.2 module add NAMD/2.12-nompi #Execute NAMD namd2 +setcpuaffinity +p28 +idlepoll +devices $CUDA_VISIBLE_DEVICES config_file > output_file
Here the user can execute several tasks on each node:
#!/bin/bash #SBATCH -A SNICXXXX-Y-ZZ #Asking for 10 min. #SBATCH -t 00:10:00 #Number of nodes #SBATCH -N 2 #Ask for 56 processes (2x28 cores on the nodes) #SBATCH -n 56 #SBATCH --exclusive #Load modules necessary for running NAMD module add GCC/6.3.0-2.27 OpenMPI/2.0.2 module add NAMD/2.12-mpi #Execute NAMD srun namd2 +setcpuaffinity config_file > output_file
The option +setcpuaffinity in these examples is used to set the affinity of the working threads which results in an increase of performance > 20% w.r.t. to the timings where that option is not used.
We evaluated the performance of the two versions of NAMD installed on Kebnekaise for a 23074 atoms system in 15,000 time steps simulation to give you an idea of the scaling behavior of this software:
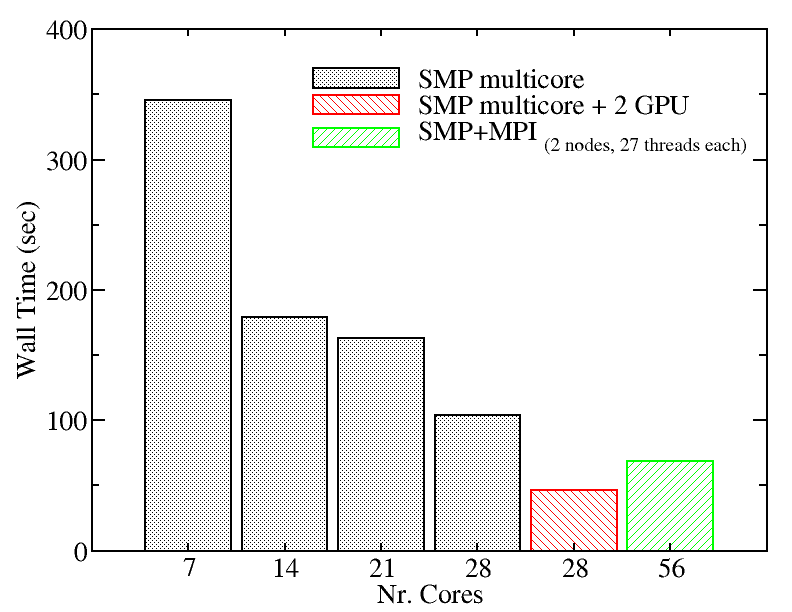
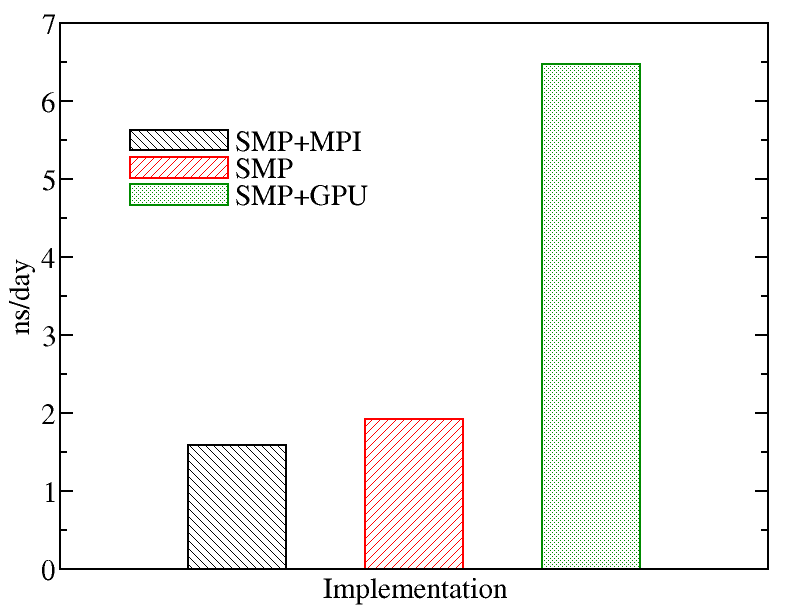
We also tried a larger system to monitor the performance of NAMD.We evaluated the performance of different NAMD implementations including SMP+MPI (with 28 cores), SMP (with 28 threads), and SMP+GPU (with 2 GPU cards). The figure below shows the best performance of NAMD. The benchmark case consisted of 158944 particles, using 1 fs. for time step and a cutoff of 1.2 nm. for real space electrostatics calculations. Particle mesh Ewald was used to solve long-range electrostatic interactions. As a comparison, we used the same test case for Gromacs (see Gromacs page).
The steps that should be followed in order to use a multi-node GPU version of NAMD can be found here.
Documentation is available on the NAMD Online Reference page.